Dossier réalisé d’après l’intervention de mesdames ETIENNE JULAN et BIBRAC du centre de drépanocytose au lycée de Pointe Noire en décembre 2004 par Dominique LEPORT
Plan :
La Maladie:
La synthèse de l’hémoglobine :
- Structure de l’hémoglobine Les gènes de l’hémoglobine
- Les différents génotypes: (les 16 premiers codonssontreprésentés)
Le Phénotype de la drépanocytose :
Historique :
La transmission :
Le dépistage
La Variabilité de la maladie : Interaction entre génotype, phénotype et environnement
- L’allèle muté du gène b confère un avantage sélectif à certaines populations
Plusieurs facteurs favorisent les crises drépanocytaires :
10 règles d'or pour minimiser la survenue de crises.
Les thérapies
La Maladie:
La drépanocytose est une maladie génétique autosomale récessive qui affecte l’hémoglobine des globules rouges; plus précisément les chaînes ?-globine qui sont modifiées au niveau de leur sixième acide aminé. La conséquence est la synthèse d’une hémoglobine anormale : l’hémoglobine S ou HbS. Les hématies normales sont discoïdes. Quand elles contiennent de l'HbS désoxygénée (désoxygénée car elle a libéré au profit des cellules, l’oxygène qu’elle transportait), elles s'incurvent, se déforment en forme de faucille, en cas de déshydratation ou d’hypoxie (manque d’oxygène). Dans un premier temps, cette déformation entraîne le blocage du sang dans les capillaires, la vaso-occlusion. La vaso-occlusion provoque des ischémies locales. En Guadeloupe, 11% de la population transmet la maladie (soit 50 000 transmetteurs environ) et 0,5% est drépanocytaire (1nouveau-né sur 300)
La synthèse de l’hémoglobine :
Un globule rouge possède 100 millions de molécules d’hémoglobine
La synthèse des chaînes d’hémoglobine, a et ß se fait sous le contrôle de gènes situés respectivement sur les chromosomes 16 et 11.
Structure de l’hémoglobine La molécule d’hémoglobine (Hb), chez les vertébrés, est composé de deux types de chaînes globine, de structure voisine : l’une appartient au type a, l’autre au type b. Les 4 chaînes contiennent chacune une molécule d’hème Les chaînes de globines déterminent le nom de chaque molécule d’hémoglobine. Dans l’espèce humaine, l’hémoglobine majoritaire de l’adulte, l’hémoglobine A (HbA), est constituée : - de quatre chaînes polypeptidiques : 2 chaînes a constituées de 141 acides aminés et 2 chaînes b de 146 acides aminés (a2b2). - de quatre molécules d’hème

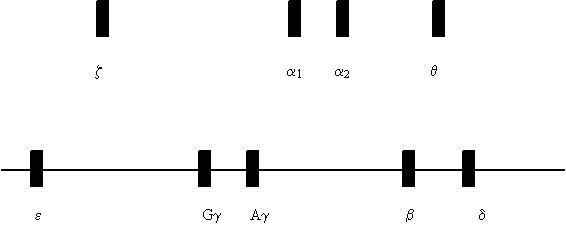
http://www.gs-im3.fr/hemoglobine/HbStructure1.html Chez l’adulte, il existe également une hémoglobine minoritaire appelée hémoglobine A2 (HbA2). Elle se distingue de l’HbA par la présence de 2 chaînes d-globine à la place des chaînes b : Hb A2 = a2d2 Les chaînes b et d globine appartiennent à la famille des chaînes de type b-globine. La synthèse des chaînes a et b-globine et celle de l’hème sont équilibrées. Synthèse de l’hémoglobine : les gènes de l’hémoglobine Les chaînes synthétisées varient en fonction du stade de développement. Le développement des techniques de biologie moléculaire et de la génétique médicale a permis d’identifier et de définir le nombre de gènes codant pour les chaînes globine, leur séquence et leur localisation sur les chromosomes chez l’Homme. On distingue les gènes globine de type a situés sur le chromosome 16 et les gènes de type b localisés sur le chromosome 11. Les chaînes a-globine sont codées par deux gènes identiques a1 et a2 situés côte à côte sur le chromosome 16. On retrouve également sur ce chromosome, d’autres gènes, de la famille des gènes a-globine, qui vont s’exprimer à des périodes différentes au cours de la vie embryonnaire et fœtale, c’est à dire au cours du développement de l’embryon puis du fœtus dans l’utérus maternel (Figures 1a et 1b, Tableau 1) L
le gène z exprimé chez l’embryon, le gène q faiblement exprimé lors du développement. Les chaînes b-globine sont codées par le gène b-globine et les chaînes d par le gène d situés sur le chromosome 11. On retrouve aussi sur le chromosome 11, les gènes e, Gg et Ag.
|
![]()
![]()
|
![]()
Figure 1a : l’ordre des gènes sur le chromosome correspond à l’ordre dans lequel ils sont exprimés au cours du développement, à partir de la conception.
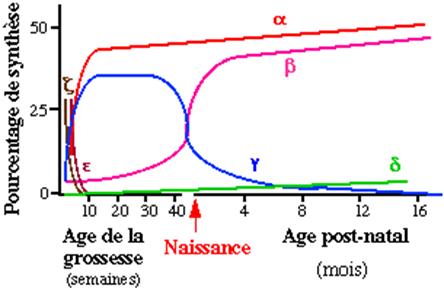
Figure 1b : synthèse des chaînes de globine au cours du développement
| Hémoglobine |
Type de chaînes |
Période de synthèse maximale |
| Hb embryonnaire | z2e2 | Avant 9 semaines de vie intra-utérine |
| Hb fœtale (HbF) | a2g2 | 9 semaines – naissance (diminue rapidement après la naissance) |
| Hb A | a2b2 | Après la naissance |
| HbA2 | a2d2 | Après la naissance |
Tableau 1 : Le profil hémoglobinique de l’adulte est atteint vers l’âge de 6 mois, chez les individus non porteurs d’hémoglobinopathies et comporte environ 97% d’HbA, = 3,1%d’HbA2 et = 1% d’HbF
Les différents génotypes : (les 16 premiers codons sont représentés)
La drépanocytose correspond à une mutation ponctuelle au niveau du sixième codon du gène b-globine : A, adénine, est remplacée par une thymine T. Cette mutation entraîne des modifications au niveau de la chaîne de ß globine, où le sixième acide aminé, un acide glutamique est remplacé par une Valine.
HbA / HbA : Individu sain
ATG GTG CTG TCT CCT GCC GAC AAG ACC AAC GTC AAG GCC GCC TGG GGC
ATG GTG CTG TCT CCT GCC GAC AAG ACC AAC GTC AAG GCC GCC TGG GGC
HbA / HbS : Individu transmetteur mais non malade
ATG GTG CTG TCT CCT GCC GAC AAG ACC AAC GTC AAG GCC GCC TGG GGC
ATG GTG CAC CTG ACT CCT GTC AAG ACC AAC GTC AAG GCC GCC TGGGGC
HbS / HbS : Individu drépanocytaire
ATG GTG CAC CTG ACT CCT GTC AAG ACC AAC GTC AAG GCC GCC TGG GGC
ATG GTG CAC CTG ACT CCT GTC AAG ACC AAC GTC AAG GCC GCC TGG GGC
HbS / HbC
ATGGTGCACCTGACTCCTGTGGAGAAGTCTGCCGTTACTGCCCTGTGGG
ATGGTGCACCTGACTCCTAAGGAGAAGTCTGCCGTTACTGCCCTGTGGG
La forme AS ne provoque pas la maladie.
Cette mutation, a présenté un avantage en protégeant ses porteurs contre le paludisme.
Exemple livre Nathan, p 132
Le gène non muté est appelé A (A étant l’initiale de adulte)
Il existe d’autre hémoglobines ß anormales liées à différentes mutation du gène A (HbC)
Les malades seront HbSS ou HbSC Les transmetteurs seront HbAS ou HbAC
Le Phénotype de la drépanocytose :
Changement de forme de la molécule d’hémoglobine :
L’acide Glutamique est chargé négativement alors que la valine est électriquement neutre, ce qui aura des conséquences sur le comportement de la molécule d’hémoglobine et de ses propriétés.
Ainsi l’HbS acquiert la possibilité de former des liaisons, des ponts entre les différentes chaînes et permettra donc une polymérisation des chaînes d’HbS.
La polymérisation se fait sous la forme désoxygénée (désoxyhémoglobine).
Cette polymérisation entraîne une perte de la forme arrondie au profit de la forme en faucille (falciforme).
DREPANO (faucille) CYTE (cellule) OSE (maladie)
La conséquence sera une vaso-occlusion.
Donc, pas de circulation du sang qui n’approvisionne plus les cellules qui vont en souffrir (la principale manifestation en est la douleur). IL s’en suit une destruction rapide des globules rouges (à durée de vie courte = 20 jours au lieu des 120 jours physiologiques)
Et une anémie (d’où le nom d’anémie falciforme donnée à la forme homozygote SS).
La rate devient défaillante et comme c’est un organe immunitaire, on observe une sensibilité accrue aux infections microbiennes. Celle-ci se manifeste par la survenue d’infections graves (septicémies ou infections généralisées, méningites, infections du rein, infection du poumon Pour lutter contre l’occlusion vasculaire, il est nécessaire de bien s’hydrater et de bien s’oxygéner.
Remarque :
La polymérisation de l’HbS est la cause majeure, mais non unique, de la vaso-occlusion. D’autres facteurs interviennent comme l’activation de la paroi des vaisseaux, des plaquettes sanguines et des lymphocytes.
Historique :
On situe l’origine de la drépanocytose à – 3000 ans, en Afrique centrale et occidentale, et dans la région arabo-indienne. A partir de ces zones d’origine, les migrations et les métissages de populations, la maladie s’est répandue dans le monde entier avec des prévalences variables d’une région à l’autre. Il s’agit de la première maladie génétique identifiée chez l’Homme.
En 1910 : Identification de la maladie
En 19.. : structure biochimique de l’hémoglobine En 1949 : Observation de l’HbS En 1959 : identification de la mutation drépanocytaire En 1960 : identification des gènes globine
La transmission :
| A | S | |
| A | AA | AS |
| S | AS | SS |
La probabilité de donner naissance à un enfant drépanocytaire est de ¼ pour les couples dont les deux membres sont porteurs du trait drépanocytaire..
Le dépistage prénatal
Dans la prévention, le diagnostic prénatal permet de rechercher le type d’hémoglobine du fœtus alors qu’il est encore dans le entre de sa mère, ceci afin, en cas de fœtus drépanocytaire, que le couple décide ou non d’interrompre la grossesse pour des raisons médicales.
Un conseil génétique, au cours duquel une information détaillée est délivrée aux couples à risque de donner naissance à un enfant drépanocytaire, permet d’aider le couple à prendre sa décision.
La drépanocytose a été la première maladie génétique identifiée.
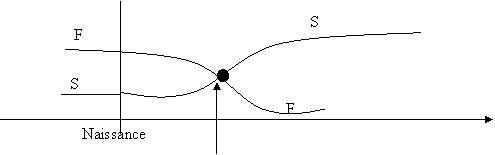 |
Evolution du rapport entre le taux d’hématies fœtales et hématies HbS
Le dépistage se fait sur les cellules du trophoblaste ou par amniocentèse, en fonction de l’âge de la grossesse. L’ADN de ces cellules est ensuite analysé (en comparaison avec celui de la mère).
Si fécondation in vitro, le diagnostic préimplantatoire est fait pour éliminer les possibles SS , mais l’échec est possible à tous les niveaux.
La Variabilité de la maladie : Interaction entre génotype, phénotype et environnement
Il existe une variabilité phénotypique de la drépanocytose, due à des facteurs génétiques et environnementaux.
Le locus de la ßglobine sur le chromosome 11 ( e ? d ß )
Dans les régions intergéniques du locus ßglobine (région du chromosome 11 qui contient l’ensemble des gènes de la famille b-globine., Des variations de la séquence nucléotidique, sans conséquence pathologique directe, sont fréquentes. Elles sont désignées sous le terme de polymorphisme. Lorsque ces modifications portent sur des sites reconnus très spécifiquement par certaines endonucléases (enzymes de restriction) elles sont faciles à mettre en évidence par les méthodes de cartographie génique (polymorphisme de taille des fragments de restriction). L'association de plusieurs de ces polymorphismes définit un haplotype.
Pour la drépanocytose, 5 types de séquences différentes autour des gènes ou haplotypes b-globine
On note 4 haplotypes africains : Centrafrique (ou Bantou), Cameroun, Sénégal et Bénin
et un haplotype arabo-indien.
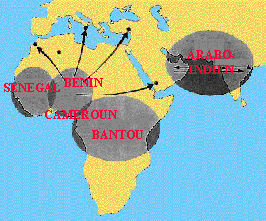
Certains haplotypes sont associés à des formes plus sévères : ex : l’haplotype centrafricain.
La forme la plus modérée est la forme arabo-indienne
L’haplotype est un facteur de la variabilité phénotypique
Ces différents haplotypes indiquent une apparition de la mutation S en cinq endroits différents simultanément.
Le taux de l’hémoglobine fœtale influence aussi la variabilité phénotypique
Plus le taux de Hb foetale est élevée, moins la maladie est prononcée.
Lors d'un effort physique important ou lors d'une exposition de l'organisme à l'altitude, l'hémoglobine se désature en oxygène, on assiste à une falciformation accélérée ce qui indique une intervention de l'environnement sur le phénotype de l'individu atteint de drépanocytose. Toute condition désaturant l'hémoglobine en oxygène est un facteur de risque de falciformation chez les sujets drépanocytaires. Les séjours en altitude sont dangereux et la pratique de sport intensif leur est interdite. Avant tout vol en avion des conseils sont donnés pour éviter des risques d'accidents
L’allèle muté du gène b confère un avantage sélectif à certaines populations
Autrefois la drépanocytose était une anomalie génétique létale responsable d'environ 100 000 morts par an. Environ 80% des homozygotes (HbS//HbS) mourraient alors avant l'âge de la reproduction. Compte-tenu d'une sélection aussi puissante contre le gène HbS, il a été longtemps difficile de comprendre pourquoi, dans certaines populations humaines, sa fréquence atteint et même dépasse largement les 10 %.
En comparant les cartes de répartition du paludisme d'une part, de la drépanocytose d'autre part, on peut remarquer leur ressemblance.
Une telle coïncidence laisse penser que l'hémoglobine HbS doit apporter un avantage dans les zones impaludées. En effet, si l'homozygote (HbS//HbS) meurt de drépanocytose, l'hétérozygote (HbA//HbS) résiste mieux au paludisme. Une explication: lorsque le parasite de la malaria s'installe dans une hématie, il en détruit l'hémoglobine; l'hématie est mal oxygénée ce qui provoque des déformations supplémentaires. L'hématie est alors détruite et avec elle le parasite qu'elle contient. Comme les hématies non parasitées sont majoritaires, le sujet survit
bien dès lors que ses parasites sont éliminés régulièrement. Les hétérozygotes (HbS//HbA) ont donc une probabilité de survie plus grande que les homozygotes (les homozygotes (HbA//HbA) meurent des suites de la malaria; les homozygotes (HbS//HbS) ne sont pas protégés).
Plusieurs facteurs favorisent les crises drépanocytaires :
La déshydratation fait perdre de l'eau au globule rouge ce qui rend le sang moins fluide.
Le ralentissement de la circulation sanguine : Tout ce qui ralentit la circulation peut créer une stase, c'est-à-dire que les globules rouges restent à un endroit et vont favoriser la crise. De nombreuses conditions ralentissent la circulation sanguine : l'effet que l'on appelle garrot (un vêtement trop serré par exemple); une mauvaise position; le froid (contracte les petits vaisseaux et ralentit la circulation); la fièvre (la déshydratation et la formation de protéines inflammatoires ralentissent la circulation); les infections (les globules blancs en excès collent aux vaisseaux et empêchent les globules rouges de circuler). Il faut donc combattre la fièvre avec des médicaments et boire beaucoup.
Surveiller sa température : Une augmentation de la température corporelle entraîne une déshydratation.
Tout ce qui fait consommer de l'oxygène en plus favorise la crise : Les efforts avec essoufflements, les efforts musculaires concentrés sur un muscle, comme l'haltérophilie, font consommer plus d'oxygène.
Tout ce qui désature l'hémoglobine en oxygène déclenche une déformation de l’hématie.
De nombreux facteurs de l'environnement agissent sur le phénotype d'un individu atteint de drépanocytose.
La vie en altitude : Au delà de 1500 m le risque augmente si on n'est pas en condition physique optimale. Mieux vaut éviter les altitudes au-delà de 2000m, contrôler les autres facteurs (froid, neige, efforts physiques).
Les voyages en avion : La pressurisation des avions correspond à une altitude de 1500 à 1800 m ce qui constitue un risque certain de crise douloureuses à cause de la baisse d'oxygène. Il faut boire abondamment, éviter la station assise prolongée, le froid et les vêtements trop serrés.
A la mer, à la piscine : attention aux écarts de températures entre l'air et l'eau, sources de crises.
L'alcool : qui est toxique est contre-indiqué chez les drépanocytaires. L'alcool déshydrate et peut déclencher des crises.
Le tabac : est très nocif pour le drépanocytaire dans la mesure où il diminue l'oxygène dans le sang.
10 règles d'or pour minimiser la survenue de crises.
1- Bien se laver le corps et les dents pour éviter les microbes provoquant des infections.
2- Surveiller sa température
3- voir un médecin.
4- Il faut beaucoup boire (environ 3 litres d'eau par jour),
5- Il faut veiller à ne jamais manquer d'oxygène donc éviter les endroits mal aérés, les hauteurs de plus de 1500 m et les voyages en avion pas ou mal pressurisés
6- Avoir une bonne alimentation, riche et variée
7- Surveiller la couleur des yeux et des urines
8- Eviter tout ce qui peut ralentir ou bloquer la circulation du sang : pas de vêtements trop serrés, de positions jambes croisées, etc.
9. Avoir un bon état dentaire
Ne jamais négliger de voir régulièrement le médecin même si tout va bien
10- Se donner et respecter une bonne hygiène de vie.
Les thérapies :
Les traitements :
Hydratation
|
Mesure quotidienne
Antalgiques
Antibiotiques
Vaccins à jour
|
|
Greffe de moelle osseuse,
Transfusion de cellules sanguines
HbSS est remplacée par HbAS ou HbAA
Thérapie génique : Clonage du gène.
Une nouvelle stratégie de thérapie génique pour traiter les hémoglobinopathies
WASHINGTON - 03/05/2000 - Une nouvelle technique de thérapie génique évaluée avec succès chez la souris pourrait permettre de traiter les hémoglobinopathies (drépanocytose, thalassémies) en introduisant l'allèle sauvage du gène directement dans les cellules hématopoïétiques. Cette technique fait appel à une présélection des cellules transfectées avant leur greffe chez le receveur
Bibliographie
La recherche et l'étude des maladies du globule rouge à Créteil
Le site sur l'hémoglobine par Henri Wajcman
Une bande dessinée
http://www.gs-im3.fr/hemoglobine/BD1.html
Nuage de mot